Our Research
This is what we are up to in the lab
Overview
Problems in protein folding are the root cause of many of the most devastating human diseases, including Alzheimer’s disease, Parkinson’s disease and ALS (Lou Gehrig’s disease).
We study the cell biology underpinning protein-misfolding diseases. Our long-term goal is to identify the critical genes and cellular pathways affected by misfolded human disease proteins.
We don't limit ourselves to one model system or experimental approach. We start with yeast, perform genetic and chemical screens, and then move to other model systems (e.g. mammalian tissue culture, mouse, fly, zebrafish) and even work with human patient samples (tissue sections, patient-derived cells, including iPS cells, and next generation sequencing approaches to look for mutations in novel genes).

Yeast models to study ALS disease protein TDP-43
We developed a yeast model to study the ALS disease protein TDP-43 (Johnson et al., Proc Natl Acad Sci USA 2008).
We have used yeast and in vitro biochemistry (in collaboration with Jim Shorter at PENN) to analyze the effects of ALS-linked TDP-43 mutations on aggregation and toxicity (Johnson et al., J Biol Chem 2009). We used these models to perform high-throughput genetic screens to elucidate the molecular pathways that regulate the function of these disease proteins and control their conversion to a pathological conformation. We are currently analyzing hits from recent high-throughput screens that identified potent modifiers of TDP-43 toxicity . We are validating these hits in cell culture, animal models (mouse, fly, and zebrafish), and human patient samples.
These TDP-43 modifier screens are providing insight in two main ways:
1. The genes and pathways that are able to modify TDP-43 toxicity in yeast are now good candidates for evaluation as genetic contributors to ALS and related disorders in humans (e.g., Elden et al., Nature 2010; see ataxin 2 below).
2. The yeast hits and their homologs are candidate therapeutic targets, especially gene deletions (Armakola et al., Nat Genet 2012; Kim et al., Nat Genet 2014).

Ataxin 2 and ALS
One of the hits from our yeast TDP-43 genetic modifier screen, PBP1, is the homolog of a human neurodegenerative disease protein, ataxin 2. We have validated this genetic interaction in the fly nervous system (in collaboration with Nancy Bonini at PENN), used biochemistry to show the proteins physically associate in an RNA-dependent manner.
We analyzed the ataxin 2 gene in 915 individuals with ALS and 980 healthy controls and found mutations in this gene as a common geneticrisk factor for ALS in humans. Long polyglutamine (polyQ) expansions (>34Q) in ataxin 2 cause spinocerebellar ataxia type 2 (SCA2). We found intermediate-length polyQ expansions in ataxin 2 (27-33Q) significantly associated with increased risk for ALS (Elden et al., Nature 2010).
Because inhibiting ataxin 2 function in yeast or fly reduces TDP-43 toxicity, we are investigating ways to disrupt the ataxin 2 / TDP-43 interaction as a potential therapeutic strategy. We found that lowering levels of ataxin 2 in mouse, either by knockout or with antisense oligonucleotides (ASOs) can markedly extend survival and reduce pathology in TDP-43 transgenic mice (Becker et al., Nature 2017). We are extending these studies to additional mouse models and testing effects of ataxin 2 lowering in human cell models.
We found that ALS cases harboring intermediate-length ataxin 2 polyQ expansions have distinct TDP-43 pathology compared to ALS cases with normal length ataxin 2 (Hart et al., Acta Neuropathol 2012). We have also found that intermediate-length, but not normal or SCA2-length ataxin 2 polyQ causes stress-induced caspase activation, TDP-43 cleavage and phosphorylation (Hart and Gitler J Neurosci 2012). Since ataxin 2 plays a key role in stress granule formation and function, we are exploring a role for ataxin 2 in integrating stress signals and how such pathways, if dysregulated, may converge in disease pathologies (Kim et al., Nat Genet 2014).
We are currently performing experiments to discover regulators of ataxin 2 function (Kim et al., Cell Rep 2022; Rodriguez et al., Cell Rep 2022) and to define the mechanisms by which polyQ expansions in ataxin 2 cause neurodegenerative diseases (Boeynaems et al., Mol Cell 2023).

Stress granules as the crucibles of ALS pathogenesis: the role of liquid-liquid phase separation
In addition to defining the role of aggregation-prone RNA-binding proteins in disease, we are actively investigating the normal cellular functions of these proteins, especially their remarkable ability to self-assemble to form functional RNA granules (e.g., P-bodies, stress granules, and neuronal RNA transport granules; Li et al., J Cell Biol 2013). We aim to define additional components of RNA granules using a combination of genetic and biochemical approaches.
We identified a novel role of the ALS disease protein profilin 1 in stress granule assembly and dynamics (Figley et al., J Neurosci 2014). Elucidating the complex cellular functions of stress granules, their constituents, and their regulators will hopefully allow us to therapeutically modulate these structures in disease situations (e.g., Kim et al., Nat Genet 2014).
Stress granules and other bimolecular condensates form by a process called liquid-liquid phase separation. We are investigating the mechanisms regulating condensate formation and the role of these membrane less organelles in normal cell biology and disease (Boeynaems et al., PNAS 2019; Dorone et al., Cell 2021; Lasker et al., Nat Commun 2022; Boeynaems et al., Mol Cell 2023).
In recent work we have used machine learning algorithms to discover a role of liquid-liquid phase separation across the tree of life, ranging from spider, scorpion, and rattlesnake venom to antimicrobial peptides secreted from immune cells (Boeynaems et al., bioRxiv 2023).

C9orf72 in FTD and ALS: Disease models and mechanisms
Mutations in the C9orf72 gene are the most common cause of ALS and frontotemporal dementia (FTD). The mutation is a massive hexanucleotide repeat (GGGGCC) expansion in the intron of C9orf72. The mechanism by which C9orf72 mutations cause disease has remained unclear and of intense interest. The nucleotide repeat is transcribed in the sense and antisense direction and these transcripts accumulate in the nucleus and cytoplasm of neurons of C9orf72 mutation carriers, where they might sequester important RNA-binding proteins and essential splicing factors. In collaboration with the Petrucelli laboratory we have recently identified a way to selectively inhibit the expression of both sense and antisense mutant C9orf72 transcripts, which could offer therapeutic potential (Kramer et al., Science 2016).
In addition to the RNA accumulating, the repeat expansion is a substrate for an unconventional form of translation: repeat-associated non-ATG translation (RAN translation), which produces dipeptide repeat proteins (DPRs) from the sense and antisense transcripts (e.g., polyGlyAla; polyGlyPro; polyProAla; polyGlyArg; polyProArg). These DPRs are themselves aggregation-prone and accumulate in the brain and spinal cord of C9orf72 mutation carriers. We have developed several new yeast models to study C9orf72 mutations. These models recapitulate salient features of the human disease, including RNA foci formation and RAN translation. The arginine-rich DPRs (GlyArg and ProArg) are particularly toxic to yeast, enabling us to perform genome wide screens for modifiers. We identified genes involved in nucleoctyoplasmic transport as potent modifiers of C9orf72 DPR toxicity in yeast and have validated these results in mammalian cells and in Drosophila (Jovičić et al., Nat Neurosci 2015; Chai and Gitler, FEMS Yeast Res 2018). These results define a pathway that may be therapeutically targeted in ALS.
We are currently using yeast and mammalian cell models to define the mechanism of RAN translation and to explore the biology of additional C9orf72 DPRs (Yamada et al., Nat Neurosci 2019). We have identified at least two genes that seem to be required for RAN translation. We hope that targeting this unconventional form of translation specifically will provide a therapeutic strategy for several neurodegenerative diseases caused by nucleotide repeat expansions (e.g., Huntington's disease and the spinocerebellar ataxias).

Next generation sequencing and single-cell transcriptomics to define ALS mechanisms
We have several ongoing projects in the lab to use exome and whole genome sequencing to define new genetic contributors to neurodegenerative diseases (Couthouis et al., PLoS Genet 2014; Cirulli et al., Science 2015). We sequenced the exomes of 47 sporadic ALS trios (ALS patient and both unaffected biological parents) to determine the role of de novo mutations in ALS. These studies suggest a potential role of chromatin regulator genes, including the neuronal chromatin remodeling complex (nBAF) component SS18L1/CREST in ALS (Chesi et al., Nat Neurosci 2013).
We have several active collaborations with neurologists at Stanford Hospital and the Veterans Administration Hospital in Palo Alto to define new disease genes for inherited neuromuscular and neurodegenerative diseases (Couthouis et al., Neuromuscul Disord 2014; Raphael et al., Brain Res 2014; Couthouis et al., PLoS Genet 2014).
We are using single-cell transcrptomics to explore the heterogeneity of the spinal cord (Blum et al., Nat Neurosci 2021) and to discover the changes during ALS, which will provide insight into mechanisms of degeneration, molecular underpinnings of selective vulnerability, and may suggest novel therapeutic strategies (Blum and Gitler, Trends Genet 2022). These studies have revealed unexpected diversity in the autonomic nervous system, gradients of fast-and slow-firing motor neuron types within motor pools, and a new subtype of skeletal motor neuron, which we have named gamma* (pronounced gamma star). We are committed to making our data and code freely available to the community (http://spinalcordatlas.org).
We have also been performing single-cell transcriptomics on human spinal cord, revealing similar logic as in mouse motor neurons, as well as novel human specific features (Gautier et al., Neuron 2023).

Genomewide screens in human cells to define mechanisms of neurodegenerative disease
We are using CRISPR/Cas9 and genome-editing to define neurodegenerative disease mechanisms in vitro and in vivo. We have initiated a large program in the lab to use CRISPR/Cas9 to perform genome wide screens in human cells for disease modifiers, starting with C9orf72 and extending to other neurodegenerative disease genes (Kramer et al., Nat Genet 2018), including TDP-43, FUS, and TBK1 in ALS, and alpha-synuclein and LRRK2 in Parkinson’s disease.
We have also used ATAC-seq to perform genome wide epigenetic analyses in primary neurons and have uncovered a surprising role of the tumor suppressor p53 in mediating neurodegeneration caused by C9orf72 dipeptide repeat protein poly(PR) (Maor-Nof et al., Cell 2021).
Discovery of novel TDP-43 splicing targets: the Achilles’ heel for FTD/ALS
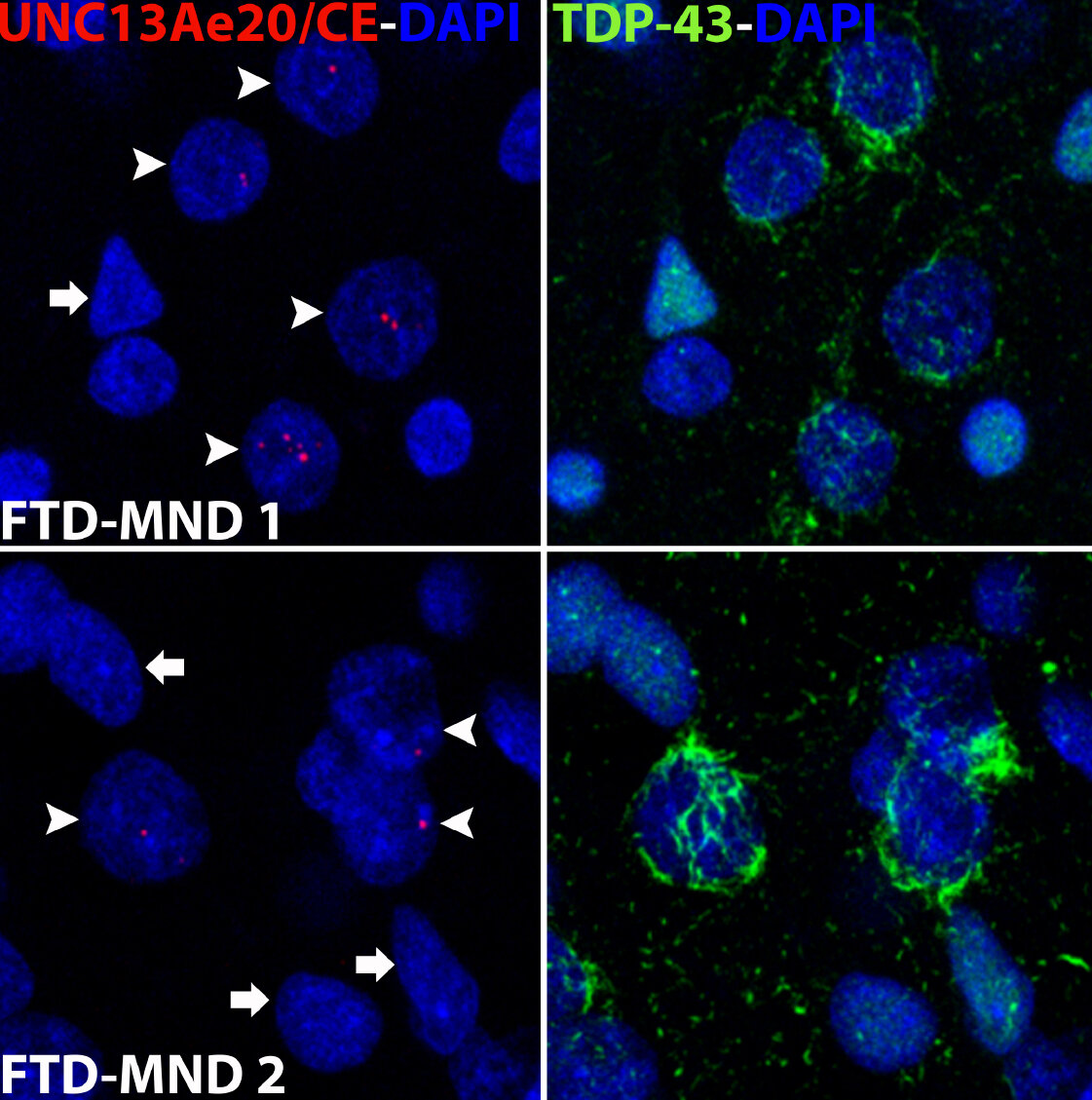
A hallmark pathological feature of ALS and FTD is the depletion of RNA-binding protein TDP-43 from the nucleus of neurons in the brain and spinal cord. A major function of TDP-43 is as a repressor of cryptic exon inclusion during RNA splicing. Single nucleotide polymorphisms (SNPs) in UNC13A are among the strongest genome-wide association study (GWAS) hits associated with FTD/ALS in humans, but how those variants increase risk for disease is unknown. We have been systematically identifying cryptic splicing targets regulated by TDP-43 in human brain. We recently discovered that TDP-43 represses a cryptic exon splicing event in UNC13A. Loss of TDP-43 from the nucleus in human brain, neuronal cell lines, and iPSC-derived motor neurons resulted in the inclusion of a cryptic exon in UNC13A mRNA and reduced UNC13A protein expression. Remarkably, the top variants associated with FTD/ALS risk in humans are located in the cryptic exon harboring intron itself and we found that they increase UNC13A cryptic exon splicing in the face of TDP-43 dysfunction. Together, our data provide a direct functional link between one of the strongest genetic risk factors for FTD/ALS (UNC13A genetic variants) and loss of TDP-43 function (Ma et al., Nature 2022).
We are currently exploring the function of UNC13A in ALS/FTD and characterizing several other novel cryptic splicing targets. Some of these represent powerful biomarkers and other ones might be therapeutic targets. We are also using genome wide approaches to identify genes that work with TDP-43 to regulate cryptic splicing.
In addition to cryptic splicing, we have also discovered loss of TDP-43 in FTD/ALS leads to widespread alternative polyadenylation changes, impacting expression of disease-relevant genes and providing evidence that alternative polyadenylation is a new facet of TDP-43 pathology (Zeng et al., bioRxiv 2024).
